Degeneration Given a Voice: How Trauma Activates the Silent History Written in Your Spine
How a motor vehicle collision can activate decades of silent disc wear—and why “pre-existing degeneration” does not mean your injury isn’t real.
Introduction: The Scan That Changed the Story
I’ve sat across from patients who hand me their MRI report with a particular expression—something between confusion and defeat. The radiologist found disc degeneration. Narrowing at C5-6. A small herniation at L4-5. Mild spondylosis throughout the cervical spine.
The insurance adjuster’s letter arrived the same week. It said, essentially: the imaging shows pre-existing conditions. Your current complaints are attributable to pre-existing degenerative disease, not the accident.
I know what you’re thinking if you’ve received a letter like that: Maybe they’re right. Maybe this was there before and I just didn’t know it.
Here’s what’s actually happening. The imaging is telling the truth. The degeneration is real, and in many cases, it was there before the accident. But the insurance adjuster’s interpretation of what that means is wrong—and it is wrong in a way that is directly contradicted by the science.
I have watched this argument be used to deny care to people who are genuinely injured. The logic sounds reasonable on the surface. The consequences for patients who accept it are serious.
This essay is about the relationship between pre-existing degeneration and acute trauma. About why disc wear that caused no symptoms for decades can become the source of significant, disabling pain after a collision. About what it means, biomechanically and clinically, for trauma to “give voice” to a pathology that was previously silent. And about why recognizing this interplay is not a matter of legal strategy—it is a matter of basic science.
Section 1: What Degeneration Actually Is — The Spine’s Normal History
To understand how trauma gives degeneration a voice, we first need to understand what degeneration actually is—and to clear away some of the fear that the word tends to generate.
Intervertebral disc degeneration is not a disease. It is not, in most cases, the result of injury or misuse. It is a normal part of the biological aging process, as predictable and universal as gray hair or reduced skin elasticity.
The intervertebral disc has a challenging anatomy. It is a largely avascular structure: its central nucleus pulposus and much of its outer annulus fibrosus receive nutrition not through direct blood supply but through diffusion from adjacent vertebral endplates. This limited nutritional access means that disc cells—the chondrocyte-like cells that maintain the disc’s extracellular matrix—are metabolically stressed throughout adult life. As early as the third decade, the water content of the nucleus pulposus begins to decline. The proteoglycan matrix that gives the disc its hydrostatic pressure and shock-absorbing capacity gradually degrades. The disc loses height. Annular fibers develop micro-fissures.
By the fourth decade, radiographically visible degenerative changes are present in a significant proportion of the population. By the sixth decade, they are the norm, not the exception.
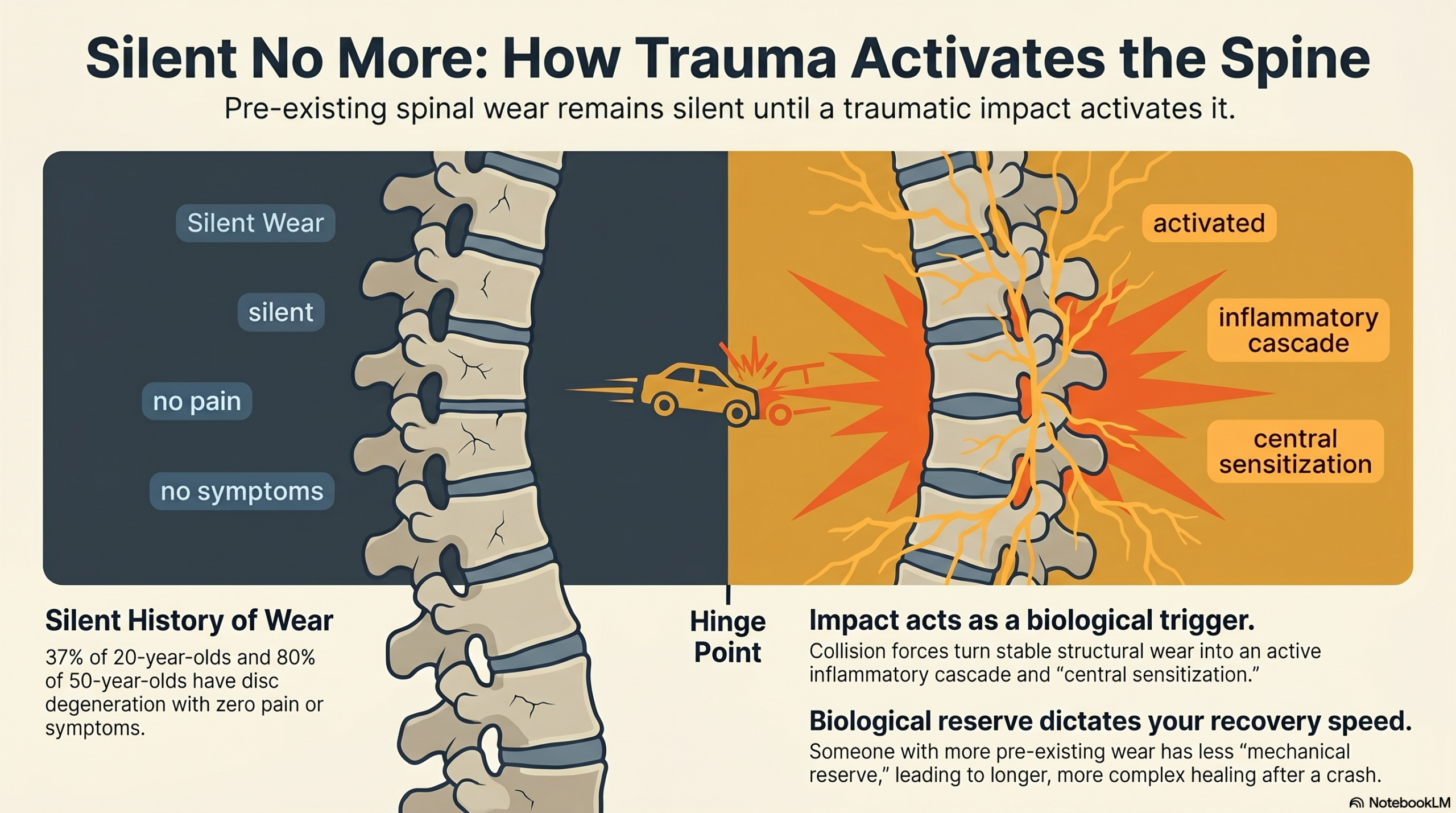
The landmark study by Brinjikji et al. (2015), published in the American Journal of Neuroradiology, performed a systematic review of MRI findings in asymptomatic individuals—people with no back or neck pain. The findings were striking: disc degeneration was present in 37% of 20-year-olds, 52% of 30-year-olds, 68% of 40-year-olds, 80% of 50-year-olds, and 88% of 60-year-olds. Disc bulges were present in 30% of 20-year-olds and 84% of 80-year-olds. Disc herniations in 29% of 20-year-olds.
None of these individuals had symptoms. The imaging findings were incidental—present, real, and clinically silent.
This is the first crucial point: degeneration and pain are not the same thing. The presence of degenerative findings on imaging does not, by itself, establish that those findings are causing the patient’s symptoms. And the absence of degeneration does not establish that no injury has occurred.
The majority of people with imaging-confirmed disc degeneration have no pain. Degenerative findings are a description of anatomy—not a diagnosis of the cause of symptoms.

Section 2: Silent Degeneration — Why Worn Discs Don’t Always Hurt
If degeneration is so common, why don’t more people have chronic pain? The answer lies in the distinction between structural change and clinical expression—between what the imaging shows and what the nervous system registers.
A degenerated disc is structurally altered. Its height is reduced. Its hydrostatic pressure is lower. Its annular fibers may have developed fissures. But these structural changes do not automatically produce pain, because pain requires nociceptive activation—the firing of pain-sensing nerve fibers—and the degenerated disc may sit in a state of chronic structural change without generating sufficient nociceptive activity to reach conscious awareness.
Several factors contribute to this quiescence.
First, the disc itself has limited innervation in healthy tissue. The outer annular fibers receive innervation from the sinuvertebral nerve and the gray rami communicantes. The inner annulus and nucleus are largely aneural. In a stable, unperturbed degenerative state, the nociceptive fibers in the outer annulus may not be sufficiently activated to generate pain.
Second, the adjacent tissues may have adapted over years to the altered mechanics of the degenerated segment. The paraspinal muscles may have developed compensatory activation patterns that reduce abnormal segmental motion. The facet joints may have remodeled to accommodate altered load distribution. The neural control system may have recalibrated its proprioceptive processing to account for changed tissue properties. The system is compromised, but it is stable—in equilibrium.
Third, the broader pain-processing system—including central pain thresholds, descending inhibitory pathways, and cognitive-emotional modulation of pain—may be providing sufficient top-down suppression of the low-level nociceptive signals the degenerated structures are generating.
The result is a spine that carries significant structural pathology while the person who owns it experiences no pain. They go to work. They exercise. They live their lives. The degeneration is silent.
Until something changes.
“The story was being written in the dark. Decades of biological change, below the threshold of awareness. Until something changed.”
Section 3: The Voice — How Trauma Activates a Previously Silent Pathology
In a motor vehicle collision, the degenerated spine faces a mechanical challenge that the adapted, compensated system has not prepared for.
The forces of a rear-end impact load the cervical spine through the S-curve whiplash mechanism we have discussed in previous writing. But for the spine carrying pre-existing degenerative changes, these forces interact with a tissue environment that is already compromised. The degenerated disc is less able to absorb and distribute compressive and tensile forces. Its reduced hydrostatic pressure means it cannot deform and recover in the normal shock-absorbing manner. Its annular fissures represent zones of structural weakness through which acute loading can propagate. The adjacent facet joints, already carrying abnormal loads due to disc height loss, are subjected to the rapid, involuntary compression of the whiplash event in a state of reduced mechanical reserve.
Several biological mechanisms then activate the previously silent pathology.
Inflammatory activation. Acute injury to the disc and adjacent structures triggers an inflammatory response mediated by cytokines including interleukin-1β, tumor necrosis factor-alpha, and matrix metalloproteinases. These inflammatory mediators do not remain confined to the injury site. They diffuse through the degenerated disc tissue, reaching the outer annular fibers and the adjacent epidural space. Research by Olmarker, Rydevik, and others has demonstrated that nucleus pulposus material—exposed through annular fissures—has direct neurotoxic and nociceptive-sensitizing effects on adjacent nerve roots. The inflammatory environment essentially lowers the activation threshold of nociceptive fibers that were previously not generating pain signals.
Neoinnervation. Chronic disc degeneration is associated with a process of pathological neoinnervation: the ingrowth of new nerve fibers into regions of the disc that are normally aneural. Studies by Freemont and colleagues demonstrated that in degenerated discs, nerve fibers—including substance P-containing nociceptive fibers—can penetrate into the inner annulus and even the nucleus pulposus. These newly innervated regions are now capable of generating pain signals in response to mechanical stimulation that previously produced no nociceptive response. Acute trauma, by loading these newly innervated regions, can activate nociceptors that did not exist—or were not functional—in the same disc years earlier.
Facet joint sensitization. The facet joints adjacent to a degenerated disc segment are carrying elevated loads due to loss of disc height. Acute whiplash trauma subjects these already stressed facet joints to rapid compressive and rotational forces. The facet joint capsule is richly innervated with mechanoreceptors and nociceptors. Injury to the capsule—even at the micro-level that produces no visible structural damage on imaging—can generate sustained nociceptive input that drives central sensitization.
Central sensitization cascade. As described in our previous essay on whiplash neuroscience, the persistent nociceptive barrage from acutely sensitized peripheral tissues can initiate central sensitization: a state in which the spinal cord and brain amplify their response to pain signals. This central amplification can make the previously subclinical nociceptive activity from degenerative structures now exceed the threshold for conscious pain perception.
This is what it means for trauma to give degeneration a voice. The structural pathology was present. The voice—the clinical expression as conscious pain, disability, and suffering—required a triggering event to emerge. The collision was that event. Not the cause of the degeneration, but unquestionably the cause of its clinical activation.
The collision does not create new pathology from nothing. It takes the silent biological history of the degenerated spine and activates it—triggering inflammatory cascades, sensitizing nociceptors, and initiating a central pain amplification process that gives clinical voice to what was previously silent.

Section 4: The Same Crash, Radically Different Outcomes
Two people in the same vehicle. Same impact speed. Same collision mechanics. One recovers in six weeks. The other is still in pain two years later.
This variation mystifies patients, frustrates insurers, and is sometimes used to imply that the slower-recovering patient’s ongoing symptoms must be psychological or volitional. The science tells a different story.
The variation in outcome after whiplash injury is one of the most studied phenomena in musculoskeletal medicine. And pre-existing degeneration—its extent, its location, and its degree of latent nociceptive potential—is one of the most significant biological contributors to that variation.
A person in their mid-thirties with no cervical degeneration, healthy disc height, intact annular fibers, and robust deep stabilizer function will absorb the whiplash event through a mechanically and neurologically intact system. Microtrauma may occur, but the nociceptive barrage is relatively contained. The proprioceptive system recovers relatively quickly. Central sensitization, if it occurs, does not have a chronic peripheral nociceptive driver sustaining it.
A person in their late forties or fifties with multi-level cervical degeneration, narrowed disc spaces, osteophytes, and reduced facet joint cartilage faces a different situation entirely. The same impact forces are distributed through a mechanically compromised system. The neoinnervated degenerated discs generate significant acute nociceptive activity. The facet joints, already stressed by disc height loss, sustain more significant capsular injury. The inflammatory mediators released acutely diffuse through tissue that is already primed for sensitization. The central sensitization cascade—once initiated—has a persistent peripheral input sustaining it.
Age itself is not the determinant. The determinant is the state of the spine’s mechanical and nociceptive reserve at the time of impact. Research by Matsumoto and colleagues showed that cervical foraminal stenosis—a common consequence of disc degeneration—was significantly associated with prolonged symptom duration after whiplash injury. The stenotic foramen has reduced tolerance for the edema that follows acute injury; the nerve root becomes compressed in an environment that has no room for inflammatory swelling.
Genetics contribute. Disc degeneration has a significant heritable component; twin studies by Battié and colleagues showed that genetic factors account for approximately 60–70% of the variance in disc degeneration, with physical loading and lifestyle factors playing a smaller role than most people assume. Two people of similar age and activity level may have radically different degrees of underlying degeneration based on factors entirely outside their control.
Psychological factors contribute—not by causing the pain, but by modulating the central processing of nociceptive input. Pre-existing anxiety, depression, or high stress can lower central pain thresholds and amplify the sensitization cascade, not because the person is “making it up” but because of well-documented neurobiological interactions between emotional regulation systems and pain-processing networks.
The patient who does not recover in the expected timeline is not weak, not dishonest, and not simply the victim of pre-existing disease that the accident happened to coincide with. They are a person whose biological reserve—shaped by genetics, aging, and the cumulative history of their spine—provided less protection against the specific event that occurred. And they deserve the same quality of care and the same legal recognition of causation as someone whose imaging was pristine.

Section 5: The Legal and Clinical Problem — Using Degeneration to Dismiss Injury
The argument that pre-existing degeneration explains away post-collision symptoms is one of the most common and most scientifically unsupportable defenses in personal injury medicine.
Its logic runs as follows: the patient has degenerative findings on imaging. Degenerative disease is a common condition that causes pain. Therefore, the patient’s current pain is caused by their pre-existing degenerative disease, not by the accident.
There are at least three fundamental errors in this reasoning.
The first error is the assumption of prior symptoms. As the Brinjikji data makes clear, the majority of people with imaging-confirmed degeneration have no symptoms. The presence of degenerative findings on a post-collision MRI does not establish that those findings were symptomatic before the collision. In the absence of documented pre-collision symptoms—medical records showing treatment for neck or back pain, prior imaging demonstrating the same findings, disability records or prior complaints—there is no scientific basis for assuming the degeneration was clinically active before the accident.
The second error is the omission of the triggering mechanism. Even if the degeneration was present before the accident, the relevant question is not whether the degenerative substrate existed but whether the collision was the precipitating cause of the patient’s current clinical state. A person may carry a genetic predisposition to inflammatory arthritis for decades without symptoms; a joint infection triggers the clinical expression of that predisposition. No one would argue that the infection was not the cause of the arthritis flare simply because the genetic predisposition existed before the infection. The same logic applies to trauma-activated disc disease.
The legal doctrine of the “eggshell plaintiff” or “thin skull rule” exists precisely to address this situation. Under this doctrine, a defendant takes their victim as they find them. A person who is more vulnerable to injury because of pre-existing conditions is entitled to full compensation for the injuries they suffered, even if a person without those conditions would have recovered more quickly. The pre-existing degeneration does not reduce the defendant’s liability; it is part of the context in which the injury occurred.
The third error is the conflation of imaging findings with causation. Imaging demonstrates anatomy. It does not establish the timeline of symptom onset or the causal relationship between structural findings and reported symptoms. A post-collision MRI showing degeneration cannot distinguish between degeneration that was present and symptomatic before the collision, degeneration that was present but asymptomatic before the collision and activated by the collision, and degeneration that has been accelerated or worsened by the collision. Clinical history, temporal relationship between the collision and symptom onset, and the specific character of the post-collision symptoms are all necessary for causation analysis. Imaging alone is insufficient.
Clinically, the consequence of accepting the degeneration-dismissal argument is that patients with the greatest biological vulnerability—the older patients, the patients with more significant baseline spine changes—receive the least support precisely when they need the most care. This is the inverse of what evidence-based medicine would recommend.
“The eggshell plaintiff rule exists for this reason: a defendant takes their victim as they find them. Pre-existing vulnerability does not reduce liability. It is the context in which the injury occurred.”
Section 6: What Appropriate Care for the Activated Degenerative Spine Looks Like
Treating a patient whose pre-existing degeneration has been activated by trauma requires understanding both the degenerative substrate and the acute injury layered on top of it.
The acute inflammatory phase requires management of the tissue-level inflammation that is driving nociceptive activation. Anti-inflammatory approaches—manual therapy directed at reducing facet joint irritation, gentle mobilization to prevent the secondary stiffening from protective guarding, ice and ergonomic modification to reduce ongoing mechanical provocation—address the peripheral drivers of the sensitization cascade. The goal is to reduce the nociceptive input that is maintaining central sensitization before the central sensitization entrenches.
Pain neuroscience education is particularly important for this patient population. Patients with visible degeneration on imaging often arrive having been told, directly or implicitly, that their spine is severely damaged and that they should expect to be limited. This catastrophic framing—which is not supported by the evidence—increases pain-related fear, drives avoidance behavior, and amplifies central sensitization. Education that accurately contextualizes degenerative findings—explaining that most degeneration is part of normal aging, that the collision activated it rather than created it, and that recovery is achievable—can meaningfully reduce fear-avoidance and improve outcomes.
Motor control rehabilitation addresses the neuromuscular consequences of both the degeneration and the acute injury. The degenerated segment has altered proprioceptive output—mechanoreceptors in degenerated facet cartilage provide less accurate feedback to the neural control system. Superimposed on this baseline deficit, the acute injury has further disrupted feedforward activation and deep stabilizer recruitment. Rehabilitation must address both layers: restoring the precision of deep stabilizer function and rebuilding the proprioceptive accuracy needed for normal segmental control.
Load management is more nuanced for the activated degenerative spine than for a purely acute injury. The degenerative segment has reduced mechanical reserve—less disc height, less hydrostatic shock absorption, smaller margin before abnormal segmental motion occurs under load. Rehabilitation must use lower initial loads, longer progressions, and more careful monitoring of pain response than might be appropriate for a younger patient with the same acute presentation. But the fundamental principle holds: the spine needs progressive load to build tolerance, and avoidance will worsen both the degeneration and the neuromuscular deconditioning.
Expectation management is clinical skill as much as science. The patient whose degeneration has been activated by trauma faces a different recovery trajectory than the younger patient with minimal baseline degeneration. The timeline is longer. The plateau may be lower. The risk of chronic symptoms is higher. Communicating this honestly—while also communicating that meaningful improvement is achievable and that the goal of care is function and quality of life, not perfect imaging—allows the patient to engage with rehabilitation realistically rather than either catastrophizing or being blindsided when recovery is slower than they were promised.
Conclusion: The Voice Was Always There. The Injury Gave It Language.
A spine that carries thirty years of degenerative change is a spine that has been living a silent biological story. The water content of the discs has been declining. The annular fibers have been developing their micro-fissures. New nerve fibers have been growing into territory they were never meant to occupy. The facet cartilage has been thinning. The mechanical reserve has been quietly eroding.
None of this produced pain. None of it demanded attention. The story was being written in the dark.
A collision changes that. It does not write a new story. It does not manufacture pathology. But it takes the silent story—the accumulated biological history of a spine that has been aging in the usual way—and gives it a voice. The inflammatory cascade amplifies the nociceptive signals that the degenerated tissues were producing below threshold. The central sensitization system amplifies those signals further. The clinical expression—pain, restriction, disability, the daily negotiation with a body that no longer feels reliable—emerges not from nothing but from a biological substrate that was waiting for a triggering event.
Two people in the same crash. One recovers in six weeks. The other carries the consequences for years. The difference is not willpower or dishonesty or the severity of the impact. The difference is biological reserve—the specific, individual history of a spine that was written long before either person ever got into a car.
Recognizing this is not a legal argument. It is a scientific one. Pre-existing degeneration does not reduce the causal significance of the collision. It explains why the collision had the consequences it had. It is the context in which the injury occurred—and context is not exculpatory. Context is part of the truth.
The voice was always there. The injury gave it language. And the patient who now lives with that voice deserves to have it heard.
“The voice was always there. The injury gave it language. And the patient who now lives with that voice deserves to have it heard.”
